DIFFERENTIATION BETWEEN GENETIC AND ENVIRONMENTAL FACTORS IN CANCER
Family studies
The frequency with which other family members develop the same cancer can provide evidence supporting a genetic contribution. Family studies have show that, for a woman who has a first-degree relative with breast cancer, the risk that she will also develop breast cancer is between 1.5 and 3 times the risk for the general population. Similar studies in gastric cancer have shown that first-degree relatives of those with cancer of the stomach have a twofold to threefold increased risk compared with the general population.
Disease associations
Blood groups are genetically determined and therefore association of a particular blood group with a disease suggests a possible genetic contribution to the etiology. It is estimated that those with blood group A have a 20% increased risk of developing gastric cancer. Blood group A is also assciated with increased risk of developing pernicious anemia. Pernicious anemia is closely associated with chronic gastritis. It appears, however, that pernicious anemia has a seperate association with gastric cancer (3-6x increased risk)
ONCOGENES
Oncogenes are the altered forms of normal genes --proto-oncogenes-- that have key roles in cell growth and differentiation pathways. In normal mammalian cells are sequences of DNA that are homokogous to viral oncogenes; these are named proto-oncogenes or cellular oncogenes. Although the terms proto-oncogenes and cellular oncogenes are often used interchangeable, strictly speaking proto-oncogenes is reserved for the normal gene and cellular oncogene (c-onc) refers to a mutated proto-oncogenes which has oncogenic properties such as the viral oncogenes (v-onc)
Chronic myeloid leukemia
Investigators in Philadelphia were the first to describe an abnormal chromosomes in white blood cells from patients with chronic myeloid leukemia (CML). The abnormal chromosome, referred to as the Philadelphia/Ph1 is an acquired abnormality found in blood or bone marrow cells.
 |
| Philadelphia translocation |
Burkitt Lymphoma
Chromosomal analysis has revealed the majority (90%) of affected children with Burkitt Lymphoma to have a translocation of the c-MYC oncogene from the long arm of chromosome 8 onto heavy chain Ig locus on chromosome 14. As a consequence of these translocation is MYC comes under the influence of the regulatory sequences of the respective Ig gene and is overexpressed 10x or more.
 |
| Burkitt Lymphoma translocation |
Proto-oncogenes can also be activated by the production of multiple copies of the gene or what is known as gene amplifications, a mechanism known to have survival value when cells encounter environmental stress. For example, when leukemic cells are exposed to the chemotherapeutic agent methotrexate, the cells acquire resistance to the drug by making multiple copies of the gene of for dihydrofolate reductase, the target enzyme for methotrexate.
Gene amplification can increase the number of copies of the oncogene per cell up to several hundred times, leading to greater amount of the corresponding oncoprotein. In mammals the amplified sequence of DNA in tumor cells can be recognized by the presence of small extra chromosomes known as double-minute chromosomes or homogenously staining regions of the chromosomes. Amplification is frequently seem with the MYC family of genes.
Function of oncogenes
| Signal transduction |
Types of oncogene
Growth factors
The transition of a cell from G0 to the start of the cell cycle is governed by substances called growth factors. Growth factors stimulate cells to grow by binding to growth factor receptors.
Growth factor receptors
Many oncogenes encode proteins that form growth factor receptors, with tyrosine kinase processing tyrosine kinase domains that allow cells to bypass the normal control mechanisms.
Intracellular signal transduction factors
Two different types of intracellular signal transduction factor have been identified:
- Proteins with GTPase activity. Mutation is ras genes result in increased or sustained GTPase activity, leading to unrestrained growth
- Cytoplasmic serine threonine kinases
DNA-binding nuclear proteins
Some oncogenes encode proteins that are specific transcription factors that regulate gene expression by activating or suppressing nearby DNA sequences.
Cell cycle factors
Cancer cells can increase in number by increased growth and division or accumulate through decreased cell death. Loss of factors that lead to normal programmed death (apoptosis) can result in the accumulation of cells. Activation of the bcl-2 oncogene through chromosomal rearrangements is associated with inhibition of apoptosis, leading to certain types of lymphoma.
Tumor suppressor genes
A tumor suppressor gene, is a gene that protects a cell from one step on the path to cancer. When this gene mutates to cause a loss or reduction in its function, the cell can progress to cancer, usually in combination with other genetic changes. The loss of these genes may be even more important than proto-oncogene/oncogene activation for the formation of many kinds of human cancer cells
Retinoblastoma
Retinoblastoma (Rb) is a relativelu rare, higly malignant, childhood cancer of the developing retinal cells of the eye that usually occurs before the age of 5 years. Rb can occur in either sporadically (non-hereditary) or familial, which is inherited in an autosomal dominant manner. Non-hereditary cases usually involve only one eye, whereas hereditary cases can be unilateral but are more commonly bilateral or occur in more than one site in one eye (=multifocal).
Two-hit hypothesis
In 1971 Knudson carried out an epidemiological study of a large number of cases of both types Rb and advanced a 'two-hit' hypothesis to explain the occurance of Rb. He proposed that individuals with a positive family history had inherited one non-functional gene that was present in all cells of individuals --germline mutation-- with the second gene at the same locus becoming inactivated somtically in a developing retinal cells. In contrast, in the sporadic form of Rb, two inactivating somatic mutation would need to occur independently in the same retinoblast cell.
! Although the hereditary form of Rb follows an autosomal dominant pattern iof inheritance, at the molecular level it is recessive because a tumor occurs only after loss of both alleles.
Loss of heterozygosity
Loss of heterozygosity (LOH) is a gross chromosomal event that results in loss of the entire gene and the surrounding chromosomal region. The loss of heterozygosity is a common occurrence in cancer, where it indicates the absence of a functional tumor suppressor gene in the lost region.
LOH can occur through several mechanisms, which include loss of a chromosome through mitotic non-disjunction. a deletion on the chromosome carrying the corresponding allele, or a crossover between the two homologus genes leading to homozygosity for the mutatant allele.
TP53
The TP53 gene provides instructions for making tumor protein p53. This protein acts as a tumor suppressor. The TP53 is the most frequently mutated of all the known cancer genesTP53/p53 major function is activation of apoptosis.
Li-Fraumeni syndrome
Members of families with this rare syndrome (autosomaal dominant) are highly susceptible to developing a variety of malignancies at an early age, including sarcromas, adrenal carcinomas and breast cancer. Point mutations in highly conserved regions of the TP53 gene have been identified in the germline of family members.
DNA Methylation and genomic imprinting
The methylation of DNA is an epigenetic phenomenon, it has the effect of silencing gene expression and maintaining the stability of the genome. Genomes of cancer cells are hypomethylated compared with those of normal cells. This loss of imprinting (LOI) may lead to activation of an allele that is normally silent, and hence the high expression of a product that confers advantageous cellular growth.
Just as hypomethylation may lead to activation of oncogenes, the opposite effect of hypermethylation may also give rise to an increased cancer risk, in this case through silencing of tumor suppressor genes whose normal functions include inhibition of cell growth.
Telomere length and cancer
The ends of the chromosomes are known as telomeres and they are specialized chromatin structures that have protective functions. Telomerase is an enzyme that can lengthen the telomeres in those cell in which it is expressed. Every cell division appears to result in the loss of TTAGGG repeats because conventional DNA polymerases cannot replicate a linear chromosome in its entorety, known as the 'end-replication problem'. This progressive loss of telomere length is a form of cellular clock believed to be linked to both aging and human disease. Some cancer cells express high levels of telomerase, so that cell viability is maintained.
Short telomeres are known to be a feature of the premature aging syndromes such as ataxia telangiectasia.
GENETICS OF COMMON CANCERS
Colorectal cancer
The majority of colorectal cancers are thought to develop from 'benign' adenomas. The transition from a small adenomatous polyp to an invasive cancer is thought to take between 5-10yrs. Mutations of the APC, RAS, TP53 genes and LOH accumulate during the transition from a small benign adenoma to carcinoma.
Familial adenomatous polyposis
Approx 1% of persons who develop colorectal cancer do so through inheritance of an autosomal dominant disorder known as familial adenomatous polyposis. Affected person develops numerous polyps of the large bowel, with more than 90% with FAP eventially developing bowel cancer.
Hereditary non-polyposis colorectal cancer - Lynch syndrome
A proportion of individuals with familial colonic cancer may have a small number of polyps, and the cancer occur more frequently in the proximal, or right side, of the colon. This familial cancer-predisposing syndrome is inherited as an autosomal dominant disorder and had been known as hereditary non-polyposis colrectal cancer (HNPCC)
Microsatellite instability (MSI) is the condition of genetic hypermutability that results from impaired DNA mismatch repair
MYH polyposis
In a large study of familial polyposis cases showed neither dominant inheritance nor evidence of an APC gene mutation. Of these families more than 20% were found to have mutations in the MYH gene. MYH polyposis is an autosomal recessive trait. Mutations that knock out the MYH gene, lead to defects in the base excision repair pathway; this is a form of DNA mismatch repair.
Juvenile polyposis syndrome
Juvenile polyposis syndrome is a syndrome characterized by the appearance of multiple juvenile polyps in the gastrointestinal tract. JPS present in variety of ways, including bleeding with anemia, pain, intussusception and failure to thrive. Two genes have identified as causative: SMAD4 & BMPR1A.
Cowden disease
Cowden syndrome (also known as "Multiple hamartoma syndrome") is a rare autosomal dominant inherited disorder characterized by multiple tumor-like growths called hamartomas and an increased risk of certain forms of cancer. Mutations in the tumor suppressor PTEN gene cause Cowden.
Two-hit hypothesis
 |
| Two-hit hypothesis |
! Although the hereditary form of Rb follows an autosomal dominant pattern iof inheritance, at the molecular level it is recessive because a tumor occurs only after loss of both alleles.
Loss of heterozygosity
Loss of heterozygosity (LOH) is a gross chromosomal event that results in loss of the entire gene and the surrounding chromosomal region. The loss of heterozygosity is a common occurrence in cancer, where it indicates the absence of a functional tumor suppressor gene in the lost region.
 |
| Loss of heterozygosity |
TP53
The TP53 gene provides instructions for making tumor protein p53. This protein acts as a tumor suppressor. The TP53 is the most frequently mutated of all the known cancer genesTP53/p53 major function is activation of apoptosis.
Li-Fraumeni syndrome
Members of families with this rare syndrome (autosomaal dominant) are highly susceptible to developing a variety of malignancies at an early age, including sarcromas, adrenal carcinomas and breast cancer. Point mutations in highly conserved regions of the TP53 gene have been identified in the germline of family members.
DNA Methylation and genomic imprinting
The methylation of DNA is an epigenetic phenomenon, it has the effect of silencing gene expression and maintaining the stability of the genome. Genomes of cancer cells are hypomethylated compared with those of normal cells. This loss of imprinting (LOI) may lead to activation of an allele that is normally silent, and hence the high expression of a product that confers advantageous cellular growth.
Just as hypomethylation may lead to activation of oncogenes, the opposite effect of hypermethylation may also give rise to an increased cancer risk, in this case through silencing of tumor suppressor genes whose normal functions include inhibition of cell growth.
Telomere length and cancer
The ends of the chromosomes are known as telomeres and they are specialized chromatin structures that have protective functions. Telomerase is an enzyme that can lengthen the telomeres in those cell in which it is expressed. Every cell division appears to result in the loss of TTAGGG repeats because conventional DNA polymerases cannot replicate a linear chromosome in its entorety, known as the 'end-replication problem'. This progressive loss of telomere length is a form of cellular clock believed to be linked to both aging and human disease. Some cancer cells express high levels of telomerase, so that cell viability is maintained.
Short telomeres are known to be a feature of the premature aging syndromes such as ataxia telangiectasia.
GENETICS OF COMMON CANCERS
Colorectal cancer
The majority of colorectal cancers are thought to develop from 'benign' adenomas. The transition from a small adenomatous polyp to an invasive cancer is thought to take between 5-10yrs. Mutations of the APC, RAS, TP53 genes and LOH accumulate during the transition from a small benign adenoma to carcinoma.
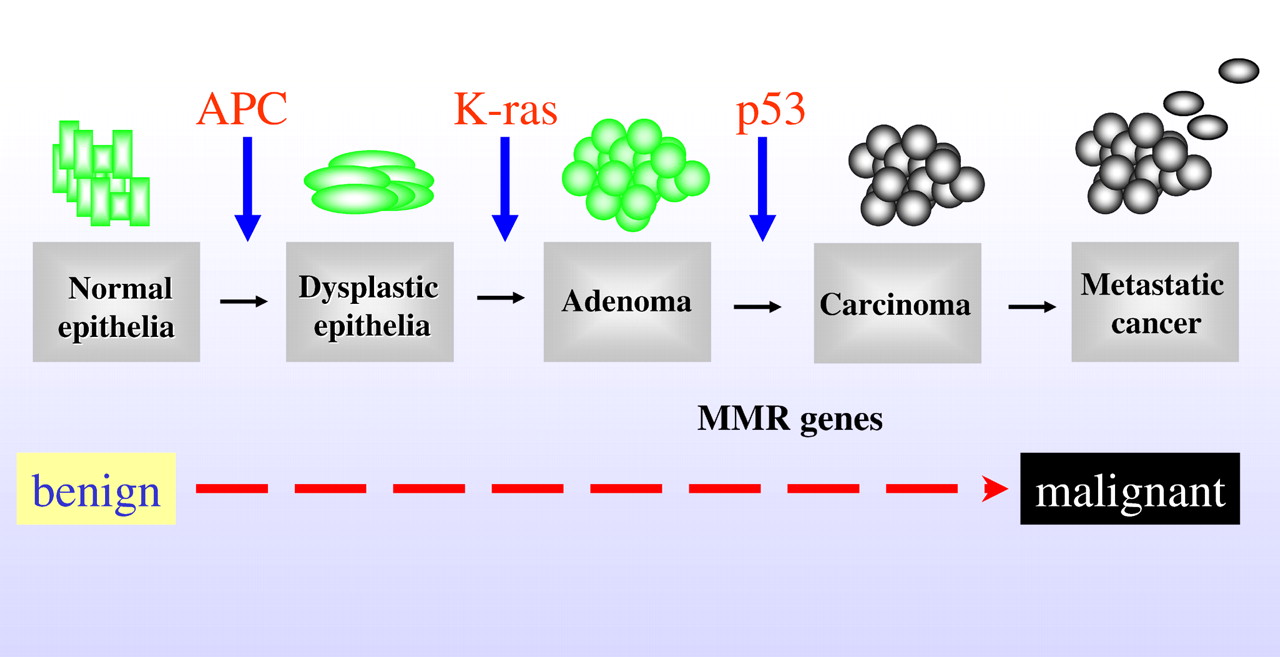 |
| Colorectal cancer multistep process |
Approx 1% of persons who develop colorectal cancer do so through inheritance of an autosomal dominant disorder known as familial adenomatous polyposis. Affected person develops numerous polyps of the large bowel, with more than 90% with FAP eventially developing bowel cancer.
Hereditary non-polyposis colorectal cancer - Lynch syndrome
A proportion of individuals with familial colonic cancer may have a small number of polyps, and the cancer occur more frequently in the proximal, or right side, of the colon. This familial cancer-predisposing syndrome is inherited as an autosomal dominant disorder and had been known as hereditary non-polyposis colrectal cancer (HNPCC)
Microsatellite instability (MSI) is the condition of genetic hypermutability that results from impaired DNA mismatch repair
MYH polyposis
In a large study of familial polyposis cases showed neither dominant inheritance nor evidence of an APC gene mutation. Of these families more than 20% were found to have mutations in the MYH gene. MYH polyposis is an autosomal recessive trait. Mutations that knock out the MYH gene, lead to defects in the base excision repair pathway; this is a form of DNA mismatch repair.
Juvenile polyposis syndrome
Juvenile polyposis syndrome is a syndrome characterized by the appearance of multiple juvenile polyps in the gastrointestinal tract. JPS present in variety of ways, including bleeding with anemia, pain, intussusception and failure to thrive. Two genes have identified as causative: SMAD4 & BMPR1A.
Cowden disease
Cowden syndrome (also known as "Multiple hamartoma syndrome") is a rare autosomal dominant inherited disorder characterized by multiple tumor-like growths called hamartomas and an increased risk of certain forms of cancer. Mutations in the tumor suppressor PTEN gene cause Cowden.
Peutz-Jegher syndrome
Peutz-Jeghers syndrome is an autosomal dominant genetic disease characterized by the development of benign hamartomatous polyps in the gastrointestinal tract and hyperpigmented macules on the lips, oral mucosa, on the palms, plantar areas and other extremities. Mutation in a serine threonine kinase gene, STK11 causes Peutz-Jegher syndrome.
Breast cancer
Family studies have shown that the risk of a woman developing breast cancer is greater when one or more of the following factors is present in the family history:
- a clustering of cases in close female relatives
- early age (<50 yrs) of presentation
- the occurence of bilateral disease
- additional occurence of ovarian cancer
One potentially key element in the development of sporadic breast/ovarian cancer is the gene named EMSY. The normal function of EMSY may be to switch off BRCA2.
BRCA1 and BRCA2 genes
Approx 40-50% of families with early onset autosomal dominant breast cancer have a mutation in the BRCA1 gene and have been shown to have a 60-85% lifetime risk of developing breast cancer. Females with BRCA1 mutation have an increased risk of developing ovarian cancer and males an increased risk of developing prostate cancer.
Mutation in the BRCA2 gene account for 30-40% of families with early onset autosomal dominant breast cancer and the lifetime risk of developing breast cancer is similar. Although breast cancer in males is very rare, males with mutations in the BRCA2 gene have a 6% lifetime risk of developing breast cancer.
Mutation in the BRCA2 gene account for 30-40% of families with early onset autosomal dominant breast cancer and the lifetime risk of developing breast cancer is similar. Although breast cancer in males is very rare, males with mutations in the BRCA2 gene have a 6% lifetime risk of developing breast cancer.
0 comments :
Post a Comment